

Treating disease rationally with antisense oligo therapy
How nucleic acids make rational drug design easier.
People in every society in history have used drugs to try to treat illness. Sometimes they were successful, but more often they were not. Until recently, the discovery of effective drugs was very slow, and those that worked were used alongside ineffective folk remedies. In truth, effective drugs were folk remedies that happened to work. By the 19th century, medicinal chemists were attempting to make drug discovery more systematic by performing experiments on animals and making rigorous observations. But they were not particularly successful, largely because the molecular underpinnings of disease were almost completely invisible to 19th century biologists.
Drug development became much more effective in the 20th century as medical scientists began to rationally design potential therapies grounded in a molecular-level understanding of disease. One of the great successes of 20th century medicine is the treatment of childhood acute lymphoblastic leukemia, which was almost 100% fatal in 1900. Millennia of human efforts to treat disease had been completely ineffective at finding a successful treatment for what we now call childhood leukemia. In the mid-20th century, Sidney Farber and his colleagues transformed the treatment of childhood leukema when they introduced folic acid antagonists as chemotherapy. This treatment was based on their growing understanding of the role of folic acid metabolism in leukemia. After decades of further improvement, nearly 90% of children diagnosed with childhood leukemia now survive.
As our understanding of the molecular basis of disease has grown, so has our ability to rationally design drugs. While we still rarely know all of the relevant molecular variables involved in any disease, we do have a significant capacity to rationally design potential therapies based on our knowledge of the proteins and other molecules that are active in pathogenic pathways. We get a lot of mileage out of a fairly simple framework: Disease can often be treated by inhibiting or activating specific macromolecules (proteins and RNA). Therefore, we can devise new treatments by designing therapeutics that physically bind to a target molecule, in order to inhibit or activate it.
It’s hard to state how profoundly different this framework is from what came before. In 1949, Linus Pauling, Harvey Itano, and Ibert Wells published a classic paper in Science, titled “Sickle Cell Anemia, a Molecular Disease.” In it they compared the chemical and physical properties of hemoglobin from patients with sickle cell disease to hemoglobin from unaffected people. They proposed, for the first time in history, an evidence-based molecular mechanism of disease, which, they said, “if it is correct, it supplies a direct link between the existence of ‘defective’ hemoglobin molecules and the pathological consequences of sickle cell disease.”
Of course, the route from an understood molecular disease mechanism to a successful cure can be a long one. Just last year, a CRISPR gene therapy for sickle cell disease became the first CRISPR-based therapy approved by the FDA, 75 years after the paper by Pauling and his colleagues. But our improving capacity for rational drug design allows scientists come up with potential therapies much faster, and enables us to treat diseases that no amount of human ingenuity could have solved under the old paradigm of drug discovery.
Rational drug design with nucleic acids
Today, rational drug design most often refers to using 3D models of protein structure to find potential surfaces to which a designed chemical or peptide inhibitor could bind. One of the most famous examples is Gleevec, a molecule that was specifically designed to inhibit the ATP binding pocket of the Abl protein kinase that drives chronic myelogenous leukemia. This kind of rational drug design is still difficult, because most proteins do not have an obvious druggable surface or binding pocket. The development of therapeutic antibodies and, more recently, AI models of protein structure do help, but the fact remains that it is very difficult to design specific inhibitors or activators of particular proteins.
But one class of therapeutics avoids the complex problem of 3D modeling altogether–DNA and RNA. Because nucleic acids interact (usually) through standard Watson-Crick base pairing, you don’t need to solve a complex 3D puzzle to design a DNA or RNA molecule that binds a specific target; all you need is the sequence of A’s, C’s, G’s, and T’s from a genome database. And because every protein that you might want to inhibit is derived translation from RNA, there is an RNA target for everything. It is completely trivial to design an RNA or DNA molecule that will bind to an RNA target. Perhaps the most striking example of the ease of RNA design is the COVID mRNA vaccine, which took just two days after the virus sequence was posted.
I don’t want to get too carried away–mRNA vaccines and oligonucleotide therapies rest on decades of work. It may be simple to design a DNA or RNA sequence, but raw DNA and RNA delivered into human tissues are not effective. They need to be chemically modified to increase their stability, reduce their toxicity, etc. It was this work that led to the 2023 Nobel Prize awarded to Katalin Karikó and Drew Weissman for their work on mRNA vaccines.
Rationally treating disease with antisense oligonucleotides
In 2017, the New England Journal of Medicine published an astonishing example of rational drug design. The study reported results of a new treatment for a neurodegenerative disease called spinal muscular atrophy (SMA). In its severe form, symptoms of motor neuron degeneration appear in infants, who are never able to sit up and eventually can’t breathe unassisted. SMA was first recognized as a distinct disease in the 1890’s, and it was invariably fatal before a child turned one or two. (There are “milder” forms of the disease with symptoms that appear in adults and become progressively worse.) In one tragic example, described in 1902, four out of eight children in the same family died of SMA:
In 1902, Beevor described a male patient with a more severe form of SMA. The patient was the eighth child in a family with eight children, and he was the fourth affected child. The first affected sibling (sister) was noticed to be paralyzed all over at the end of the first month of life and died before 4.5 months of age. The second affected sibling (sister) developed symptoms at 6 months of age, and died at 8 months of age. The third affected sibling (gender unknown) developed symptoms at 6 weeks of age and died at 7 months of age. The patient was noticed to be paralyzed all over, excepting the diaphragm, before 5 weeks of age, and died after living eight weeks.
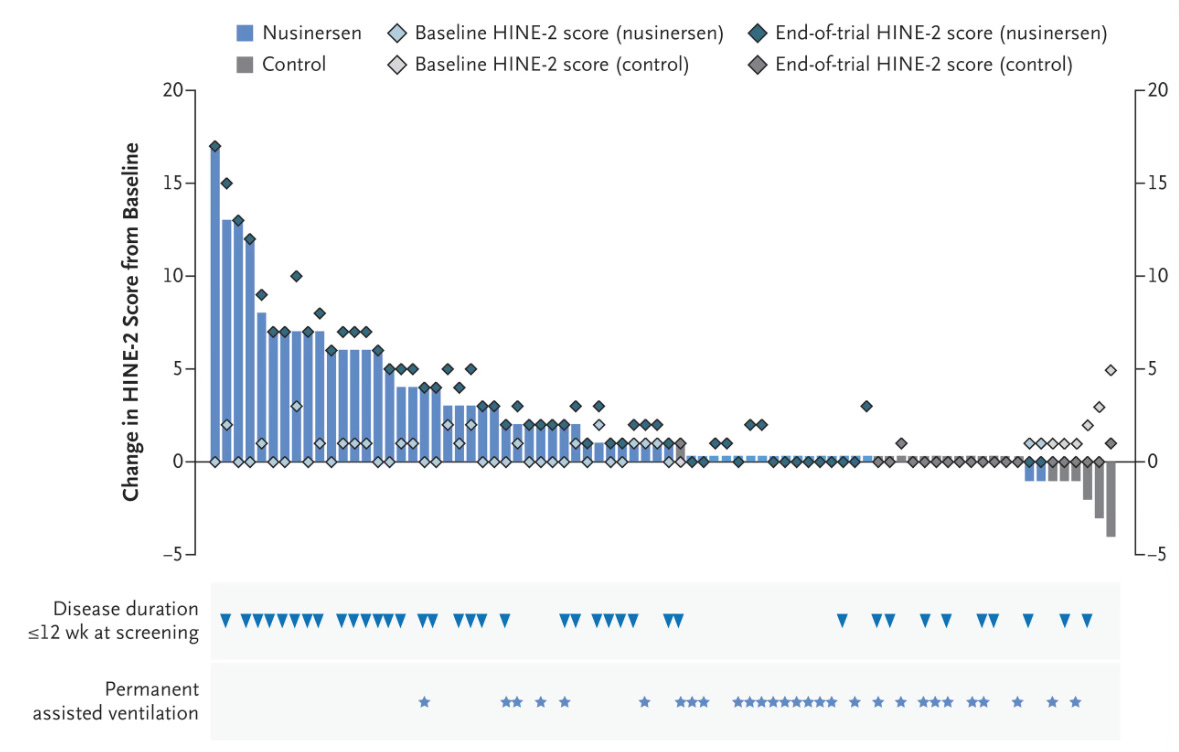
In the 2017 study, led by Richard Finkel of the Nemours Children’s Hospital in Orlando, Florida, 51% of infants treated with a new therapeutic reached a developmental milestone of motor function, compared to 0% of infants who received the placebo. The effect was so impressive that the study was terminated early, so that other children could be treated. A related study, which began treating infants with the disease before the onset of symptoms found that all 25 children in the study were alive several years later, at a median age of 4.9. The new therapeutic that successfully treated this fatal disease was nusinersen, an antisene oligonucleotide (ASO). Nusinersen was not the first ASO treatment, but it has become the most successful. The nusinersen story illustrates the power of rational nucleic acid drug design
The concept of an ASO to treat disease is simple, though development is still challenging. Thinking back on our framework for rational drug design, the goal is typically to design a drug that either inhibits or activates a molecular target. In the case of ASOs, the molecular target is the RNA transcript of a gene. The ASO is designed to have a sequence that is the reverse complement (‘antisense’) of a short region of the RNA molecule. When the ASO binds to the RNA, the RNA can be affected in several ways. The most straightforward effect is shown below: ASO-bound RNA is degraded by RNase H1, which breaks down RNA/DNA double helices.
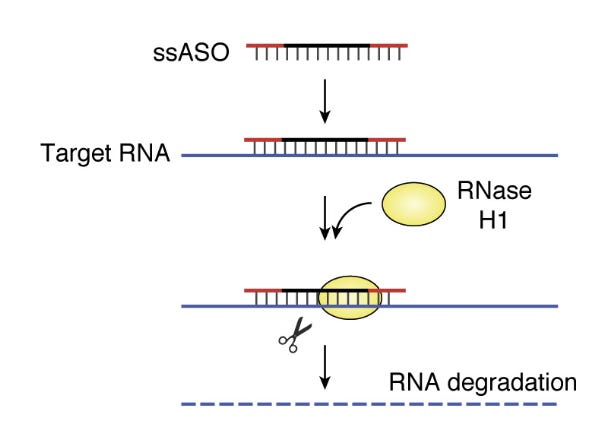
Other ways that an ASO destabilized an RNA target is prevent translation of the RNA into protein by physically blocking the binding of the ribosome, or by preventing the RNA from being properly spliced, leading to nonsense-mediated decay. You can also stabilize an RNA in various ways, including by physically obstructing a binding site for a negatively-regulating microRNA (miRNA), thereby preventing RNA degradation. This recent review has a nice discussion of different approaches. The key point is that, while a big fraction of proteins in their 3D forms might be undruggable, every messenger RNA that codes for those proteins can likely be inhibited or stabilized by an ASO.
In the case of nusinersen, a rational design was based on careful knowledge of the underlying cause of SMA. In the 1990’s scientists mapped the genetic basis of SMA to a gene called SMN1, which is necessary for the survival of motor neurons. When an infant is born with two loss-of-function mutations in SMN1, they develop the disease. However, there is a gene that can compensate for the loss of SMN1, called, sensibly enough, SMN2. The problem is that SMN2 rarely produces a functional protein, because it is spliced improperly, and one of the critical exons is skipped. In 2006, a team led by Ravindra Singh at U Mass Worcester described an ASO that that blocked the site on the RNA that caused exon-skipping. The ASO thereby led to the production of functional SMN2 protein. In patients whose SMN1 genes are non-functional, nusinersen increases the expression of functional SMN2, which compensates for the missing SMN1.
It took 10 years to go from this original ASO design to an FDA-approved drug. For a previously untreatable, fatal genetic disease, that is a fairly fast drug development timeline. Since the approval of nusinersen, gene therapy to cure the disease and a small molecule drug that has the same effect as nusinersen have been approved. That’s great news for SMA patients, and nusinersen is now being considered a bridge therapy to keep patients healthy before receiving gene therapy.
Future ASO Therapeutics
I don’t want to sound too pollyannish; identifying individuals with ASO-correctable diseases and finding RNA sites that can be effectively targeted with ASOs requires careful development. And delivery of ASOs can also be a challenge. To treat neurological diseases, ASOs, which can’t get through the blood-brain barrier, need to be injected intrathecally (into the spinal cord). SMA patients have a miracle drug available, but it requires such injections every four months.
But the tremendous advantages of rational drug design with ASOs, which now include a massive collection of biotechnological and computational tools that didn’t exist in 2006 when nusinersen was designed, has created a growing opportunity. One of these includes the ability to create organoids derived from a patient's cells, which allows scientists to rapidly screen many different ASOs for effectiveness in the lab. These new tools have created broader opportunities, and ASOs are in development to treat various cancers and other neurological diseases like Huntington’s.
One of the most interesting opportunities is the potential to treat very rare genetic diseases, even diseases for which the size of the patient population might only be 1. In a review article published last year, a group called the N=1 Collaborative, led by Timothy Yu at Boston Children’s Hospital, argued that the potential to create rationally designed, individualized therapies with of ASOs and other nucleic acids requires the medical community to think about how to evaluate treatments designed for a single person. There are thousands of extremely rare genetic diseases, which cumulatively affect a lot of people. Many of these diseases could be treated with ASOs, but there are too few individuals in each case to assemble a statistically well-powered clinical trial to evaluate the therapy. Yu and his colleagues layout a framework for developing rigorous N=1 trials.
In a sense, such efforts at individualized drug testing are a throwback to the era before rational drug design, when potential therapies were tried on small numbers of patients, and physicians would carefully follow the patients to see whether they got better or worse. Without a foundation of knowledge of the biological basis of disease, this approach wasn’t very effective. But with today’s molecular knowledge and capacity for rational drug design, we have the chance to reclaim part of that older paradigm and design afforable, individualized treatments for thousands of rare diseases.
Just want to say that I found this an interesting read (do you know you can also use them to truncate mRNA and therefore protein, restoring partial function?) Came across this while looking for grad profs, thanks for the distraction and leaving a note so I can come back after apps