

This Week’s Finds: Replication, Enhancer Testing, More AI
Five Friday links to items that caught my eye in genomics and beyond.
1. Replication initiatives conflate replication with reproducibility
I’m a little late to this, but last December Science reported on a new NIH initiative to fund contract research organizations (CROs) to replicate studies. The NIH invited researchers to apply for $50,000 to be used to help a CRO re-run their experiments. Apparently there were few takers. I think anyone who has spend time trying to get a difficult assay to work in the lab might understand the reluctance to participate. The Science article quotes one researcher who sums up the problem well:
Sean Morrison of the University of Texas Southwestern Medical Center, who edited some of the papers resulting from that project, notes the contract labs didn’t have the resources to do pilot experiments or repeat studies to work out kinks. And such labs often lack “the expertise of academic laboratories, especially when it comes to advanced or specialized techniques,” he says. In some cases, “this led to uninterpretable results.” The murky outcome of some replication attempts may have left scientists behind the original studies feeling their reputation was unfairly tarnished, he adds.
While there are some specific types of studies that should be replicated (replication cohorts for GWAS are good), I and others have been arguing for a long time that it is foolish to conflate replication with reproducibility in science. Chris Drummond of the Canadian National Research Council summed it up well in a piece written way back in 2009 (PDF):
A critical point of reproducing an experimental result is that irrelevant things are intentionally not replicated. One might say, one should replicate the result not the experiment…The sharing of all the artifacts from people’s experiments is not a trivial activity.
Cell Reports in 2014 published an example of just how hard it is to exactly replicate someone’s experiments:
Our two laboratories, one on the East and the other on the West Coast of the United States, decided to collaborate on a problem of mutual interest—namely, the heterogeneity of the human breast. Despite using seemingly identical methods, reagents, and specimens, our two laboratories quite reproducibly were unable to replicate each other’s fluorescence-activated cell sorting (FACS) profiles of primary breast cells. Frustration mounted, given that we had not found the correct answer(s), even after a year. Rather than giving up or each publishing our data without the other laboratory, we decided to work together to solve these differences, even traveling from one laboratory to the other in order to perform experiments side by side on the same human breast tissue sample. This exercise confirmed our suspicions and resolved our problem. Here, we summarize our cautionary tale and provide advice to our colleagues.
Reproducibility, I contend, is usually achieved when multiple labs that use different methods show that a phenomenon is robust. Reproducibility can be poor when journals publish papers with poor controls, p-hacking, and other bad statistical practices. The solution to that is not replication initiatives but rather holding papers to higher standards for publication.
To reiterate, there are specialized cases in biomedical science where something closer to exact replication is indeed important, but those cases generally involve studies with methods that can and should be standardized: GWAS, clinical trials, pre-clinical testing of therapeutics, etc. But exactly replicating a basic science study like this 2010 Cell paper (one included in the Cancer Reproducibility Project) is a waste of time and resources.
2. Yet another DNA language model
Yun Song and colleagues at UC Berkeley have a paper in Nature Biotechnology that describes a DNA language model trained on the multiz multi-species alignment of 100 vertebrates. What makes their model stand out, the authors claim, is the computational efficiency of the approach compared the big foundation models out there. (I covered one of them here.) Their method is efficient because they didn’t actually train their model on 100 vertebrate genomes, but rather only on the top 5% conserved regions, plus some randomly selected regions.
They tested their model by asking it to classify pathogenic variants from various databases (ClinVar, COSMIC, OMIM, etc.). Their method sometimes does about as well as, and occasionally better than some other standard variant predictors, like CADD and ESM-1b. None of the methods do particularly well on deep mutational scanning data. They scored all 9 billion possible SNVs in the human genome, and have thus created another database of predicted variant effects.
There is nothing special about the performance here, but as this field develops, it is useful to watch the performance of two different approaches to language models: multi-species models, like this one, and functional genomics models, like Enformer. It’s not clear which approach is more effective.
3. Accelerating in vivo enhancer assays
Evgeny Kvon, of the snakefied ZRS enhancer fame, has a cool new paper out describing an efficient, two-color assay to test enhancer variants in vivo:
Here we introduce dual-enSERT, a robust Cas9-based two-color fluorescent reporter system which enables rapid, quantitative comparison of enhancer allele activities in live mice in less than two weeks.
They make one-color reporter mice, each carrying either the wild-type or alternate allele of an enhancer. The mice are crossed to generate two-color offspring, which allows you to measure both versions of the enhancer in the same mouse. This promises to make in vivo enhancer assays much more efficient and reproducible.
4. Training data for deep learning beyond the genome
I don’t intend to use too much of this space to discuss my own work, but we have a paper out in Cell Systems this week that, imho, works on an angle in genomics and machine learning that I think is a under-appreciated. The paper is the thesis work of Ryan Friedman, a fantastic former graduate student who is now a postdoc in Cole Trapnell’s lab. Ryan used a technique called active machine learning to iteratively train models of regulatory DNA on successive rounds of functional assays performed in the mouse retina:
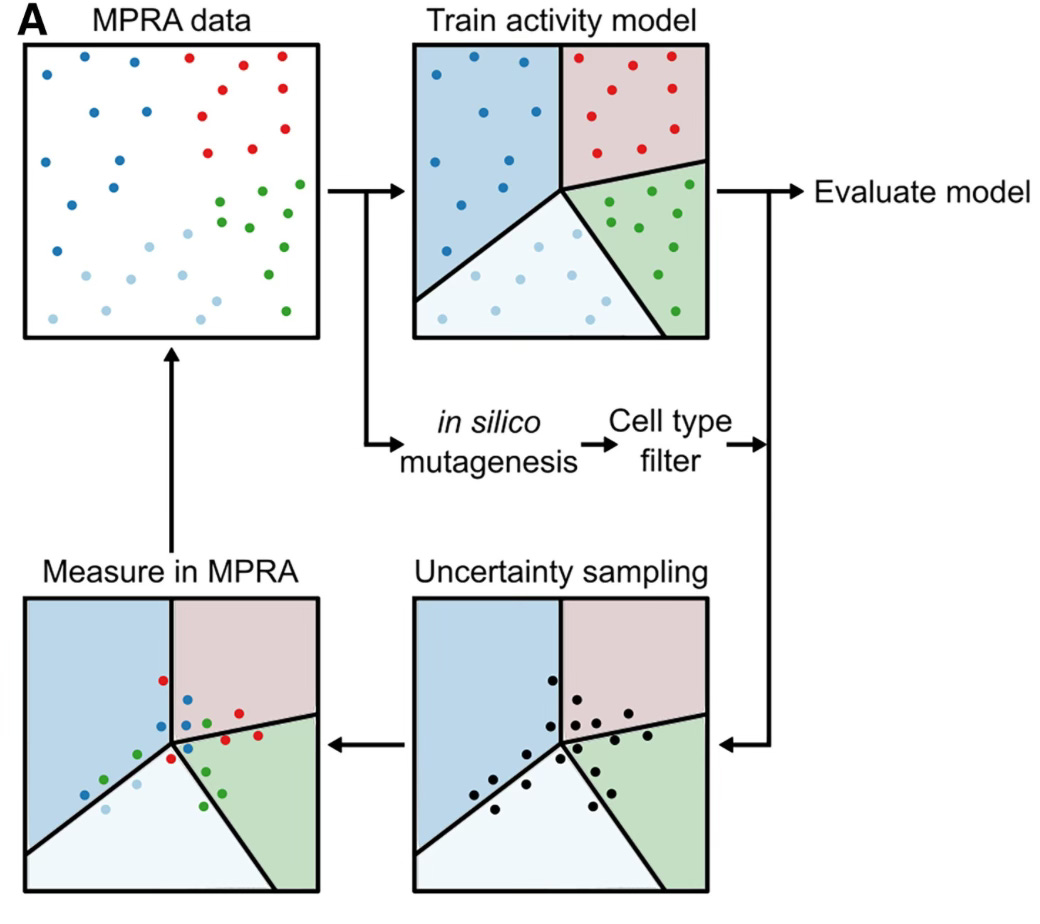
An important point of this approach is that, with functional assays and affordable DNA synthesis, we aren’t limited to training data from the genome. In fact, we can design our experiments to produce training data that is better optimized for deep learning. That the genome may not offer the best training data is something that is increasingly recognized in the field — see this commentary (“Hold out the genome”) by De Boer and Taipale (bioRxiv here), and this very good review by La Fleur, Shi, and Seelig. There isn’t enough sequence diversity in the genome to learn some of the things we want to learn, and a big chunk of the genome is inactive and probably not especially informative. Obviously not all (or even most) genomic assays use synthesized DNA, but we have a number of powerful functional assays that do, and we should take advantage of the opportunity to design experiments to better achieve our deep learning goals.
5. Is it harder to make scientific progress?
Now and again people write big think pieces asking whether scientific progress is slowing. I don’t think you should generalize. AI and biology seem to be firing on all cylinders right now. But perhaps things are different in physics. Physicist Chad Orzel, who write Counting Atoms made the point in a way I found interesting. If you consider that huge progress in physics has been made by finding anomalies, like the Michealson-Morley experiment that failed to detect the movement of the earth, or precession of Mercury that could not be accounted for by classical physics, then this point seems important:
All the anomalies in the sixth decimal place were measured a century ago— modern physicists are looking for changes in the 14th decimal place. You can still come up with new ideas about physics, sure, but they’re incredibly tightly constrained by prior experiments, and testing those theories is vastly more complicated an expensive than it was in the golden era— you used to be able to discover new particles by leaving some photographic plates out on top of a mountain, but now you need millions to billions of dollars of sophisticated electronic detectors.
“All the anomalies in the sixth decimal place were measured a century ago.” Science is cumulative in an important sense, and the scale at which we need to search to find new phenomena in some fields becomes increasingly harder to achieve, because all of the relevant measurements at the easier scales have been made. Biology is not quite the same, but there are some parallels. Samples sizes of GWAS need to be very large to detect the associations that we’re going after these days. Bulk tissue measurements of gene expression aren’t enough; we now need single-cell resolution, and maybe some spatial information too. There are still big questions to answer, but smart people have been at this business for awhile, always pushing at the technological frontier.