

How to train your model
...when you don't have enough data. A new preprint presents a clever simulation.
It’s probably not controversial to say that the limiting resource for better AI tools in biomedical research is not GPUs or new AI architectures, but data. As a general rule, more training data is needed to build more capable models. The problem is that there may not be enough data in the world to achieve the capabilities we want from AI. This paper predicts that the biggest large language models (LLMs) could run out of human text data as early as 2026. And then there are specific AI applications, like reading CT scans or tissue sections, which (at least for now) rely heavily on supervised training that requires human-labeled data (images) that the model learns. Labeling images to create these datasets requires time-consuming and expensive human effort.
An obvious solution to the data problem is to simulate training examples. With a good simulation, you could generate as much training data as you need. How do you create a good simulation? For LLMs like GPT-4, you could use the trained model to generate much more text, and then train the next on the generated text, etc. That kind of recursive training risks sending your model into a strange place. A paper published last year reported that AI models collapse when trained on data they generated. The authors wrote that “the model becomes poisoned with its own projection of reality.”
An alternative to recursive training is to simulate your data with a different model, one that is not tied into deep learning model that you are attempting to train. My WashU Genetics colleague Willie Buchser and his lab and collaborators at the McDonnell Genome Institute (MGI) have a preprint out that uses an interesting approach: they use the popular 3D computer graphics software Blender to simulate biophysically realistic tissue samples. They then use the simulated data to train a convolutional neural network (CNN) that learns to automatically annotate real tissue samples.
Using all of the data in spatial ‘omics
The context in which Willie and his colleagues are trying to automate tissue annotation is the exploding new field of spatial ‘omics. The ability to measure the expression of all genes in cells and bulk tissues was transformative back when RNA-seq arrived in the scene in 2008. But tissues are composed of complex arrangements of cells of different types, and even cells of the same type may be in different gene expression states at different locations in a tissue. The invention of technologies to perform RNA-seq on single cells was an important step forward because now different cell types in a tissue could be distinguished based on their gene expression profiles. But while single-cell RNA-seq resolves the identity of individual cells, information about the spatial distribution of these cells within a tissue or a tumor is lost.
Barely a decade after the introduction of RNA-seq, RNA sequencing within intact tissues arrived, known as spatial transcriptomics. Science advances in large part by the ability to make measurements at new scales, and the capacity to determine cell types and their expression states on the spatial scale of whole tissues is beginning to transform disease genomics.
A detailed discussion of these technologies will be a post for another day. In the context of the new work by Willie and his colleagues, an important advantage of spatial transcriptomics is that it provides not just gene expression values and spatial coordinates, but actual images of tissue slices whose features can be annotated with labels that mark known anatomical features. But this anatomical information isn’t often used because most spatial ‘omics analyses take what the authors call a bottom-up approach. When working from the bottom up, the molecular sequencing data is used to group individual cells by their gene expression patterns, and then only afterwards are cell clusters mapped back onto spatial coordinates. Instead of directly using prior information about anatomical structure of the tissue, anatomical regions are roughly reconstructed by the clusters of cell types mapped back on to the tissue coordinates. The MGI group argues instead for a top-down approach, in which cells are first labeled by the annotated anatomical region from which they came, after which gene expression patterns can be compared:
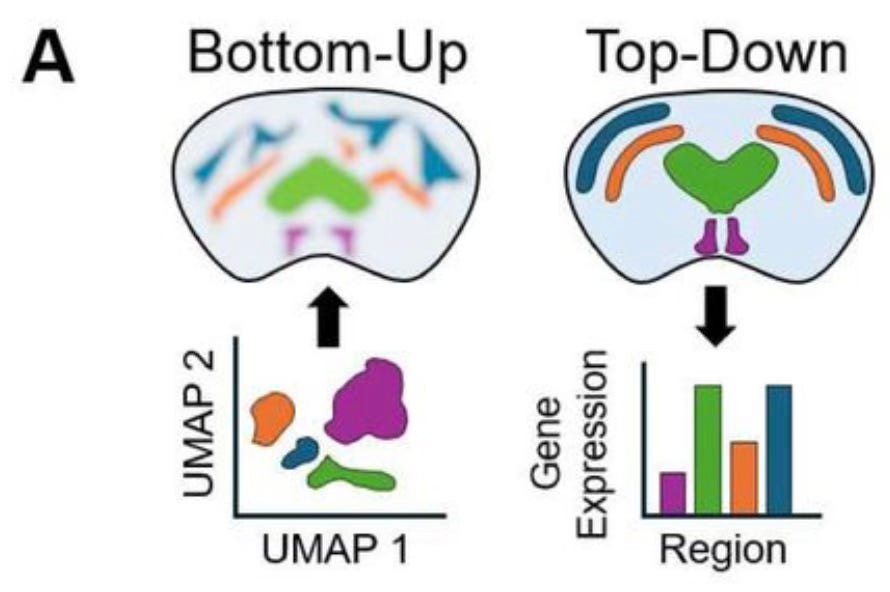
Automation is great but annotation is expensive
The top-down approach would have many important applications, such as comparing healthy and pathological brain samples. But the limitation is that annotating tissue sections is time-consuming and requires a pathologist or some other expert to manually mark the boundaries of different regions on each new image. This is just not feasible in most settings, which is why the top-down approach isn’t widely used. But the result is that, as the MGI group points out, information in the dataset is left on the table.
Software that could automatically segment tissue images into different anatomical regions would make the top-down approach feasible. Image analysis is a task that neural networks excel at, so such automatic segmentation of tissue images would be a natural job for a CNN. This, however, takes us back to the data problem I mentioned at the outset: obtaining labeled data to train neural networks is expensive, and in the case of tissue annotation, the data doesn’t exist at the scale you’d need to train a sufficiently good model.
The solution that Willie and his colleagues came up with is the main point of this post: they developed a method to simulate realistic images of tissue sections, and then used the simulated data to train a CNN capable of automatically segmenting anatomical regions in images of tissue slices. Here is the outline of how it works:
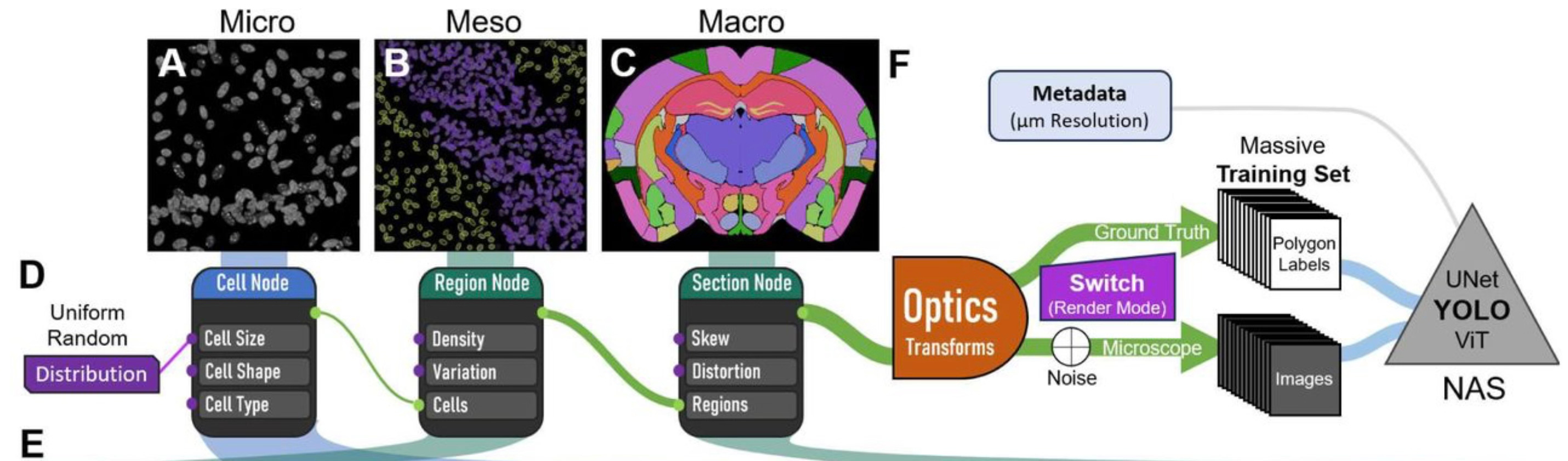
They being by simulating individual cells with different sizes, shapes, and intensities, determined by the simulated cell type. This microscale simulation is then used to build a regional simulation, in which populations of cells are built up into larger regions based on simulation parameters for cell density and variation in cell types. Regions are then built up into whole tissue sections, such as the simulated coronal section of a mouse brain shown in panel C above. All of this is done with the CAD software Blender, an open-source suite of tools used for game and movie animations.
Once the 3D tissue simulations are put together, the MGI group then simulates the process of microscopic imaging, including adding debris, tears, optical distortions, etc.:
To do this, we use optics and the rendering engine within Blender. Specifically, we employ a virtual camera with a 15.5 mm focal length and a low f-stop (0.01) to focus on the tissue section from about 1.4 mm away. Then the rendering engine ‘Cycles’ calculates the light being emitted by or bouncing off the samples and uses it to produce an image. After the image layer is produced, we use a compositor layer to add varying amounts of white noise (0-95%), making the image slightly harder to interpret. This process is repeated (per ‘frame’) for the total number of images and produces the simulated ‘microscope’ images for our dataset.
The simulated images are then used to train the segmentation CNN, which they call Si-Do-La (Simulate Don’t Label). Using a few desktop computers, they generated ~4000 simulated images per hour.
How well does Si-Do-La predict the boundaries of cells and anatomical regions? For the detailed performance assessment, check out the paper, but the authors show that Si-Do-La performs well at different scales on samples of mouse brain and spinal cord, and pig sciatic nerve. Here is an example from the paper:
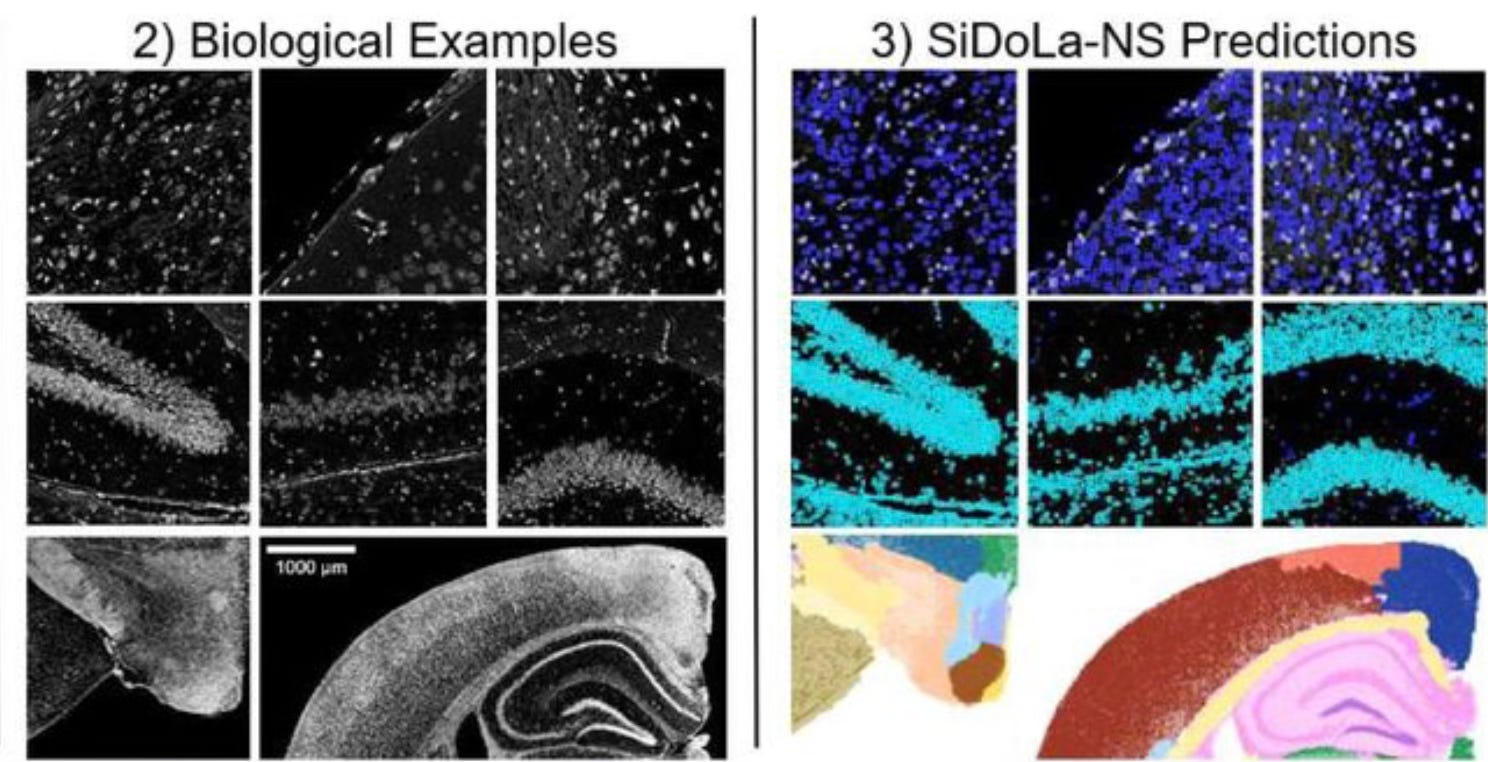
I like this paper because it is a clever example of a realistic simulation that is independent of the machine learning model that will be trained on the output. Unlike recursive training, this approach avoids the risk that the model will fall ever deeper into its own mistaken projection of reality. Realistic simulations probably can’t be done to address every type of data shortage, but it will be worthwhile to put effort into developing good simulations where they are feasible. This might include genome sequence, since the human genome itself is too small and lacks the sequence diversity we’ll need to achieve many of our modeling goals. (Though not all… see AlphaFold.) Simulating data is only one tool in the toolbox when it comes to dealing with limited training data, but there is surely more potential in simulations that could be realized with clever methods like the one described by my MGI colleagues.
Awesome, I didn't understand the paper when I first went through it but your post clears up a lot of confusion. interested to see how you think if something like this simulation -> training can be applied to functional genomics, especially in sparse single cell data?
It seems like the strength here was a great mathematical model to simulate images, is this the first challenge?