

A single-cell panorama of selective neuron failure in neurodegenerative disease
A new paper looks at transcription networks that explain why some types of neurons fail and others don't.
Coronary brain section through the hippocampus, Mikael Häggström and brainmaps.org under CCA 3.0
{kind=link}
Earlier this week I pointed to the McDonnell Genome Institute’s idea for a ‘Resiliency Project’, a large-scale effort to understand why only certain neuron types fail in particular neurodegenerative diseases. The puzzle is why, in different neurodegenerative diseases, some types of neurons fail while other do not, and why the types that fail differ among diseases. A new paper in this week’s issue of Cell addresses the same theme, and it demonstrates the remarkable power of single cell technologies to get at questions that were essentially impossible to address only a decade ago.
The work comes out of Daniel Geschwind’s lab at UCLA. His group profiled post-mortem brain samples from a set of patients affected by one of three types of neurodegenerative disease, Alzheimer’s disease (AD), frontotemporal dementia (FTD), and progressive supranuclear palsy (PSP). All three are characterized by tau pathology, which occurs when misfolded tau protein accumulates into insoluble aggregates in the brain. What causes tau pathology, and to what degree tau accumulation is a cause or effect of neurodegeneration is not well understood. But what is clear is that in each of these diseases, distinct regions of the brain are affected. This is what is meant by selective resiliency: in one disease, one set of neurons and glial cells are fail, while in another, those same neurons and glial cells are unaffected. The mystery is why.
The Geschwind lab set out to examine differences and commonalities among AD, FTD, and PSP at cellular resolution, using single-nucleus RNA-seq and single-nucleus ATAC-seq, two methods that in the past decade have made it possible to profile individual cellular states in a complex tissue like the brain. These technologies have produced astonishingly detailed new views of the cellular composition of the brain at scale. It’s now possible to profile the transcriptional and epigenetic states of thousands or tens of thousands of individual cells from a tissue, and thereby learn features of the subcellular biology of the brain that could never be seen under even the most powerful microscope. Before single cell sequencing technologies came online, we were limited to low-resolution profiles that came from heterogeneous bulk tissue samples (all of the individual cell states were averaged out), or to immortalized cell lines that don’t much resemble what’s going on in an intact brain. Using single cell technologies, the Geschwind lab was able to catalog depleted cell types and altered transcriptional network states in the different samples. They were thereby able to discover shared and disease-specific pathways and cell types that were altered in the three conditions.
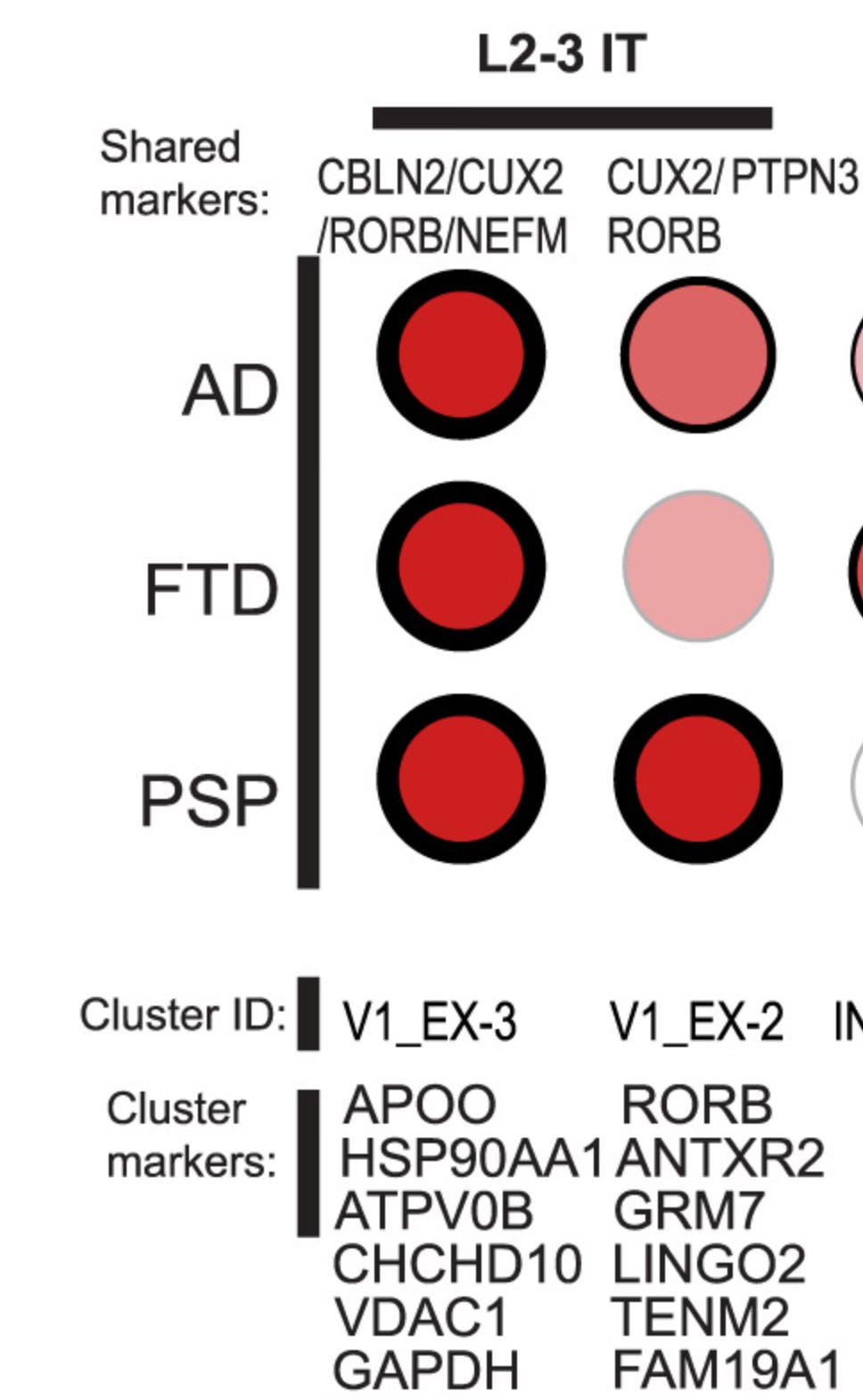
Shared and disorder-specific cellular state changes. Excerpt from Fig. 2b of Rexach, et al Cell 2024 S0092-8674.
So what did they find? It’s a dense, data-rich paper, and there are an enormous number of cell type × cell state × disease condition combinations to look at. Here are a few examples of the kinds of findings highlighted in the paper:
Among depleted neurons there were both common and disorder-specific markers (i.e., expressed genes). For example KCNH7 (a potassium-gated voltage channel) marked vulnerable neurons in all three disorders, and the authors suggest that this gene could be a potential therapeutic target. In other cases a gene that was normally expressed at low levels (RORB) in layer 2/3 neurons, was upregulated in these neurons in FTD, and these were vulnerable in FTD. Excitatory neurons in layer 5 marked by RORB were depleted in AD, but not in the other diseases.
In each condition, there were disorder-specific depletions of particular sets of neurons. For example in PSP, a layer 5/6 population of near-projecting neurons were selectively depleted, and these neurons may normally innervate nearby brain regions that have high tau pathology in PSP.
A gene regulatory network (involving the genes MAFG and NFE2L1) was anti-correlated with tau pathology, suggesting that it might be protective. Downstream genes in this regulatory network include some that are known to be neuroprotective. Notably, neurons depleted in PSP lost the expression of this regulatory network.
It goes on like this - a wealth of single-cell-resolution detail that is meaningful to neurobiologists who are deeply familiar with the various brain cell types affected by these diseases, and a bit of a slog for the rest of us. The big picture is this: A high-resolution, cross-disease comparison of the internal states of brain cell types creates a rich, informative picture of the pathways underlying the selective resiliency of different cell types in neurodegenerative disease. Sorting out cause from consequence here will obviously take much more effort to map out, but as the authors put it, creating this comparative atlas of cellular states creates “a clearer picture of shared and disease-specific aspects of resilience and vulnerability to inform the therapeutic roadmap.” Selective resiliency is an important clue to the mechanisms underlying neurodegenerative disease, something that comes to afflict almost half of those who live past 85. Those mechanisms can be glimpsed through the window of single-cell ‘omics, which creates a rich, detailed map for those trying to find a way to treat these diseases.